摘要:
TCGA数据获取、处理和分析流程(二)
上期(TCGA基因/miRNA表达谱数据整合)使用shell 脚本对多样本表达谱文件整合,实现方式是将1881个miRNA和425个样本直接存入一个”awk二维表”然后输出,全部在内存中运行,所以速度极快。但shell中已经提供了专门整合文本的命令
join,可以用于两个文本的合并.本期介绍join命令的语法以及使用join整合miRNA表达谱数据.
先从一个例子介绍join命令。
如文件a.txt内容如下:
joe 100
jane 200
herman 300
chris 400
文件b.txt的内容如下:
joe 20
jane 10
herman 30
chris 98
a.txt和b.txt分别是是两个月的业务员的名字和销售量.我们需要合并这两个月的内容.脚本如下:
# sed '/^#/d' filename用于去掉注释行,join要求文件必须排序,所以使用sort命令,而我们需要按照两个文件的第一列合并,所以是sort -k 1,1
# 为了不产生中间文件,所以使用()建立子进程并使用<重定向.
join <(sed '/^#/d' a.txt|sort -k 1,1) <(sed '/^#/d' b.txt|sort -k 1,1)
得到的结果如下并且符合预期要求:
chris 400 98
herman 300 30
jane 200 10
joe 100 20
这里只对join命令进行简单介绍,更多语法及参数可以去
极客学院查看.文章末尾点击”阅读原文”即可获得一份通俗易懂的Linux基础教程.
下面是使用join实现上期文章)中相同的功能:整合多样本miRNA表达谱文件到一个表达量矩阵:
#########################
# 使用join函数进行合并:
bash
file_ID=(`awk '{if(NR>1)print $1}' ../gdc_manifest.2017-05-26T16-02-11.963011.tsv`)
file_name=(`awk '{if(NR>1)print $2}' ../gdc_manifest.2017-05-26T16-02-11.963011.tsv`)
# 获得文件路径数组:
for((i=0;i<${#file_ID[@]};i++)){
file_path[$i]="./"${file_ID[$i]}"/"${file_name[$i]}
echo ${file_path[$i]}
}
# 使用join尽心合并:
#复制第一个文件作为起始(结果)文件,其他文件与这个文件合并
awk 'BEGIN{OFS="\t";}{ #如果第一行是列名(用"miRNA"匹配),则直接跳过行
if(NR==1 && $1~/miRNA/)next;
print $1,$3;
}' ${file_path[0]} > ../miRNA_exp_matrix.txt #写入../miRNA_exp_matrix.txt
temp1=`mktemp -t join.XXXX` #创建临时文件temp1
temp2=`mktemp -t join.XXXX` #创建临时文件temp2
for((i=1;i<${#file_ID[@]};i++)){
awk 'BEGIN{OFS="\t";}{ #处理第一个文件,如果第一行是列名(用"miRNA"匹配),则直接跳过行
if(NR==1 && $1~/miRNA/)next;
print $1,$3; #这里根据实际情况选择,比如我的数据是第一列是基因编号,第二列是表达值,那这里$3就要改成$2
}' ${file_path[$i]} > "$temp1" #写入temp1
#合并,第一个文件的第一列(miRNA)与第二个文件第一列(miRNA)合并.
join -t $'\t' -1 1 -2 1 <(sort -k 1,1 ../miRNA_exp_matrix.txt) <(sort -k 1,1 "$temp1") >$temp2
cp $temp2 ../miRNA_exp_matrix.txt
# 临时文件至空:
cat /dev/null >$temp1
cat /dev/null >$temp2
# 显示处理的进度:
echo $i
}
# 删除临时文件:
rm $temp1 $temp2
#添加首行列名:
aaa=`echo miRNA ${file_ID[@]}|sed 's/ /\t/g' `
sed -i "1i $aaa" ../miRNA_exp_matrix.txt
}
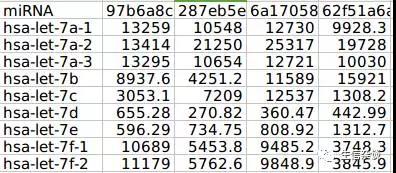
结果如下,并且与上期相同,但由于这个脚本会产生临时文件反复对硬盘读写,速度比上期的脚本明显慢了很多(慢了100倍)。当然在上面的for循环中如果将对临时文件temp1的处理放入子进程()里不产生临时文件速度会快很多。但代码看起来就太冗余了。
 苏公网安备 32050602011302号
苏公网安备 32050602011302号