本期使用TCGA的API下载肝癌的miRNA表达谱及临床数据为例说明如何下载TCGA数据.。
首先在TCGA官网的data portal界面左侧的filter的cases选项卡选择liver,files选项卡选择miRNA-seq, 数据类型选择 miRNA Expression Quantification,也可复制下面的链接打开:
https://portal.gdc.cancer.gov/search/f?facetTab=files&filters={"op":"and","content":[{"op":"in","content":{"field":"cases.project.primary_site","value":["Liver"]}},{"op":"in","content":{"field":"files.experimental_strategy","value":["miRNA-Seq"]}},{"op":"in","content":{"field":"files.data_format","value":["TSV"]}},{"op":"in","content":{"field":"files.data_type","value":["miRNA Expression Quantification"]}}]}
上面是肝癌373个cases的miRNA的miRNA表达谱链接,从url解码出来的(顺便提下后面的格式是和TCGA的API格式一样)。把链接复制地址栏打开后如下所示:
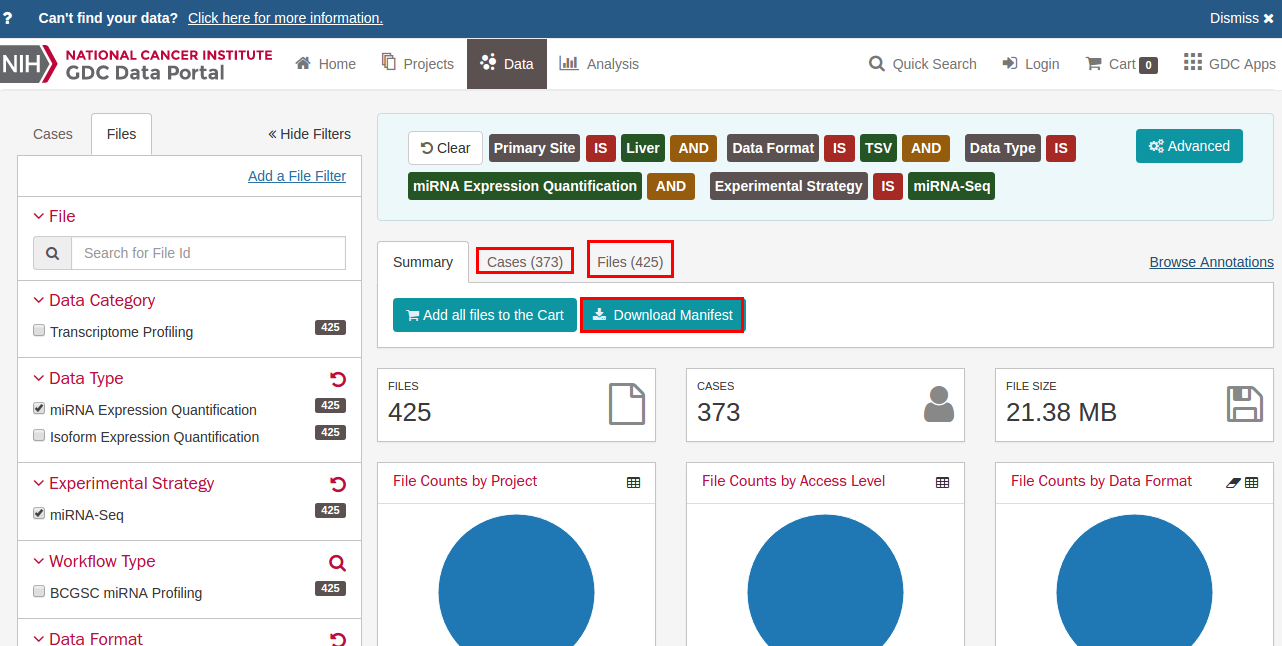
可以看出肝癌中miRNA的病例(cases)有373个,但miRNA表达谱却有425个,也就是说是有重复的,并且这425个中既有肝癌组织的,也有正常组织的.然后点击中间的Download Manifest,下载包含这425个文件详细信息的.tsv文件,文件内容如下:
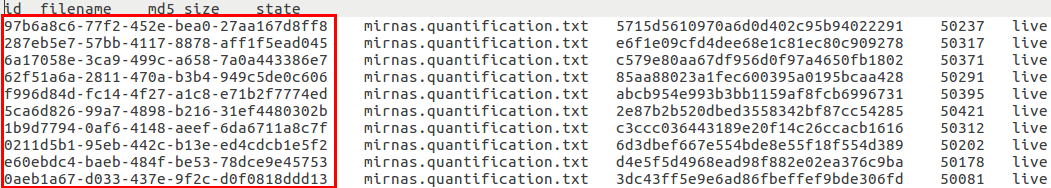
我们只需要第一列file_id,然后使用GDC Data Transfer Tool根据第一例的file_id下载这425个miRNA表达谱文件 ,Gdc工具从这里下载: https://gdc.cancer.gov/access-data/gdc-data-transfer-tool .
使用wget下载ubuntu版的:
# 下载gdc客户端:
wget -c -t 0 https://gdc.cancer.gov/files/public/file/gdc-client_v1.2.0_Ubuntu14.04_x64.zip
#解压缩出文件gdc-client
unzip ./gdc-client_v1.2.0_Ubuntu14.04_x64.zip
# 使用gdc-client根据刚才的.tsv文件下载425个miRNA表达谱文件:
mkdir miRNA_425
cd miRNA_425
#GDC添加到环境变量
sudo cp -pi ./gdc-client /usr/local/bin
使用gdc-client下载的命令格式应该是gdc-client + download + file_id:
awk '{if(NR>1){print "gdc-client download",$1}}' ../gdc_manifest.2017-05-26T16-02-11.963011.tsv | head
然后使用下面的命令进行下载,通过管道传给sh(sh命令是执行从标准输入或一个文件中读取的命令):
awk '{if(NR>1){print "gdc-client download",$1}}' ../gdc_manifest.2017-05-26T16-02-11.963011.tsv |sh
下载完成后会出现425个文件夹,以file_id命名,里面有一个mirnas.quantification.txt和logs文件夹,部分还会有个annotations文件,我们首先对下载的文件进行md5验证,看看是否全部都完整的下载了下来:
#注:由于linux系统会对所下载的文件夹按照文件名进行排序,所以进行MD5校验之前建议先把manifest中的数据按照id那一列进行排序,不然会后面校验会出现不匹配的情况而报错。
# 每个下载下来的表达谱文件都放在以file_id命名的文件夹里,通过md5验证文件的准确性:
# 将manifest文件第1列的文件id和第3列的md5值提取到数组:
file_all_md5=(`awk 'BEGIN{OFS=""}{if(NR>1)print $1,$3}' ../gdc_manifest.2017-05-26T16-02-11.963011.tsv`)
# 从下载下来的文件夹中获得所有下载下来的文件ID:
download_all=(`ls`)
# 先看数量:
echo ${#file_all_md5[@]} #425
echo ${#download_all[@]} #425
# 数量一致,然后根据md5值进行验证:
count_file=0
for file_id in ${download_all[@]}
do
# 通过md5sum命令获得每个文件的md5值:
file_id_md5=`md5sum ./$file_id/*.mirnas.*|awk '{print $1}' -`
# 判断文件md5是否与manifest文件中一致
if [[ "${file_all_md5[@]}" =~ "$file_id$file_id_md5" ]]
then
count_file=`echo $count_file + 1|bc`
else
echo $file_id"中的文件有误"
fi
done
echo "$count_file"
结果显示61755bc3-6947-4599-9f41-7d0ac7e94093中的文件有误,我看了下果然发现这个文件没有下载完全,于是使用下面的命令重新下载.哪个文件下载错误直接替换ID重新下载即可。
重新下载命令:
#download后面加下载有误的文件ID:
gdc-client download 61755bc3-6947-4599-9f41-7d0ac7e94093
# 使用md5sum重新验证这个文件,再与manifest文件比较,发现md5值一致.
md5sum ./61755bc3-6947-4599-9f41-7d0ac7e94093/mirna*
基因表达谱文件下载方法与上面类似就不再赘述.上面下载下来的miRNA表达量都是每个样本一个文件,格式如下:
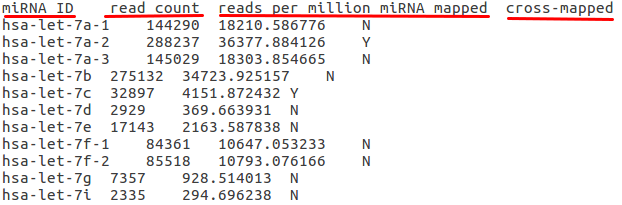
某个样本的miRNA-seq表达谱
前几列都好理解,依次是miNRA名称,原始reads数目,归一化reads数RPM,最后一列cross-mapped表示multiple-hitted miRNA,具体可以参考[PMID: 21637827 ]: miRNA cross-mapping is a prevalent phenomenon where miRNA sequence originating from one genomic region is mapped to another location
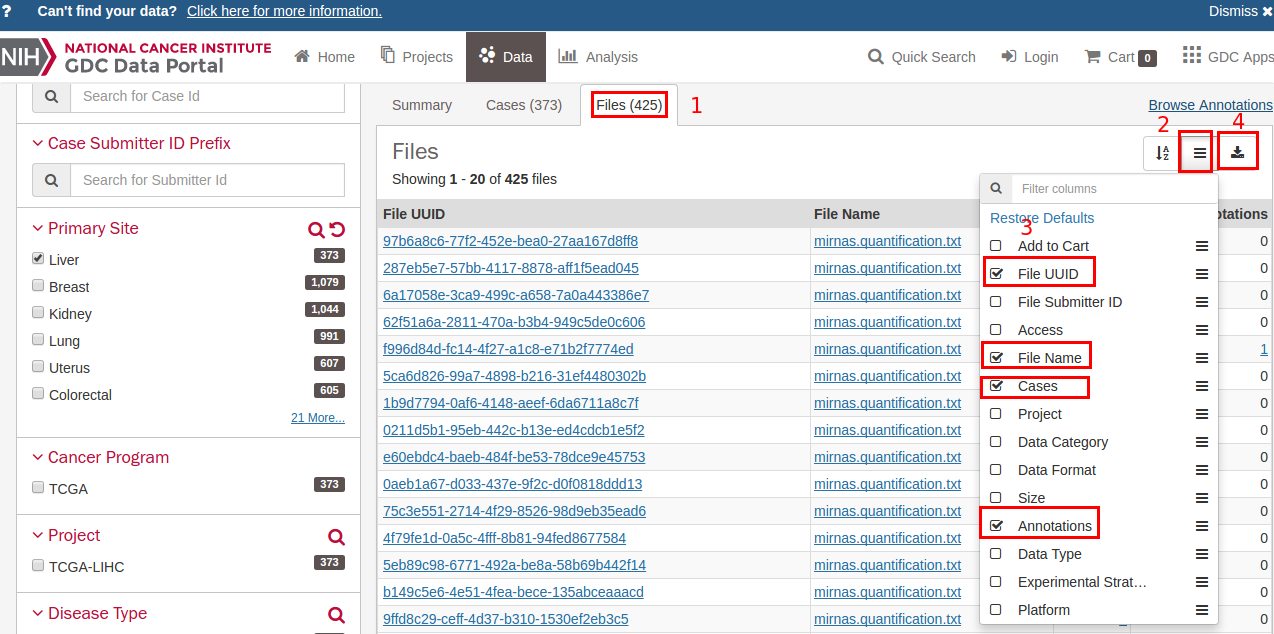
我们接下来需要看看这些样本的注释(可以用来做生存曲线),还是再刚才那个网页里,点击Files(425),然后选择要显示的列,必须选择”file_uuid”和Cases,然后点击右边的下载箭头,下载json注释文件.
下载下来的json文件是这样的,大括号{}表示对像, 用 在处理时用 点号 来引用 对象的 属性/或函数 (函数后面要加小括号). 中括号[]表示数组, 用['下标']的方式来引用. json的数据, 不管是key, 还是 value, 都要用 双引号. 表示包含425个以大括号分割的文件的文件名(file_name),病人id(case_id)和文件id(file_id):
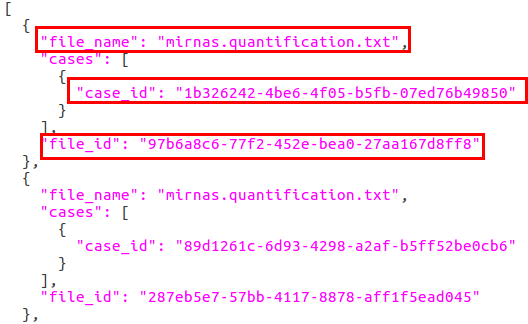
然后用awk将这425个file_id和373个case_id对应起来,顺便给个顺序ID:
# 第一列是case_id,第二列是file_id:
awk 'BEGIN{
OFS="\t";
FS="[\t\" :]+"
}
{
if($2=="case_id")
printf "%s\t",NR$3;
if($2=="file_id")
print $3
}' ../TCGA_liver_cases_detail.json | awk 'BEGIN{OFS="\t"}{print NR,$0}' - > ../miRNA_caseID_fileID
# 上面提取case_id我用的python处理,源代码处理会报错,不知道为什么会在case_id前面加编号
import json
case_list = []
with open('./files.2020-10-14.json','r',encoding='utf8') as dt:
json_data = json.load(dt)
for i in json_data:
case_list.append(i['cases'][0]['case_id'] + '\t' + i['file_id'])
f = open('case_id.txt', 'w')
for id in case_list:
f.write(str(id)+'\n')
f.close()
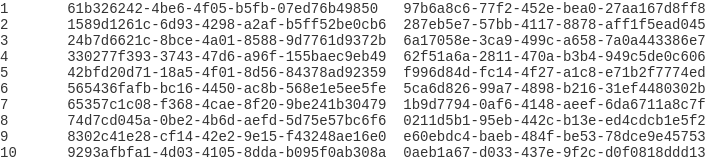
接下来使用TCGA的API获取每个case或每个样本的临床数据及注释信息,可以获得json格式的也可以获得tsv格式的,方法如下:
1. 根据case id使用TCGA 的API提取每个case的json格式的diagnoses信息(主要是days to death用于做生存曲线),获得文件cases_373_diagnoese.json:
bash
# 根据上面的case_id,使用TCGA提供的API获取cases的最新诊断信息:
# 获得所有case id并存入数组,注意加sort -u去除重复,去重复后发现只有373个case ID了,说明有4个人是有重复的.
cases_id=(`awk '{print $1}' ../miRNA_caseID_fileID|sort -u`)
# 如果没有安装jq,则sudo apt-get install jq,用于等会处理.json文件
# 创建空文件处理diagnoses数据:
cat /dev/null >../cases_373_diagnoese.json
for cid in ${cases_id[@]}
do
# 被注释这段代码可以提取制定key的值.
# echo "curl 'https://api.gdc.cancer.gov/cases/$cid?pretty=true&expand=diagnoses'" |sh|jq '.data.diagnoses[0] | {updated_datetime: .updated_datetime,tumor_stage: .tumor_stage,age_at_diagnosis: .age_at_diagnosis,vital_status:.vital_status,morphology:.morphology,days_to_death:.days_to_death}'
#提取case的diagnoses的所有key及value:
echo "curl 'https://api.gdc.cancer.gov/cases/$cid?pretty=true&expand=diagnoses'" |sh|jq '.data | {case_id,diagnoses}' |awk '{print }' >>../cases_373_diagnoese.json
done
通过上面的代码可以获得json格式的373个case的diagnoses信息,4个重复的没有显示.

373个病例的诊断信息.json格式
2. 根据case id使用TCGA 的API提取每个case的tsv格式的diagnoses信息(主要是days to death用于做生存曲线),获得文件cases_373_diagnoese.tsv:
#根据case ID创建过滤条件文件获得每个case的days to death:
cat > ../payload <<EOF
{
"filters":{
"op":"in",
"content":{
"field":"cases.case_id",
"value":[
EOF
# 将case ID写入条件:
row_num=`wc -l ../miRNA_caseID_fileID | awk '{print $1}' -`
awk -v row_num=$row_num '{if(NR<row_num)printf "\"%s\",\n",$2; if(NR==row_num)printf "\"%s\"\n",$2}' ../miRNA_caseID_fileID >> ../payload
# 创建条件结尾:
cat >> ../payload <<EOF
]
}
},
"format":"TSV",
"size":"10000",
"pretty":"true",
"expand":"diagnoses"
}
EOF
# 使用curl命令调用条件../payload获取373个cases的diagnoses:
curl --request POST --header "Content-Type: application/json" --data @../payload 'https://api.gdc.cancer.gov/cases' >../cases_373_diagnoese.tsv
#注意你的miRNA_caseID_fileID文件有几列,如果第一列是序号,则代码不变,否则,这边$2得改成你的文件中case_id对应的列号。下段代码同理。
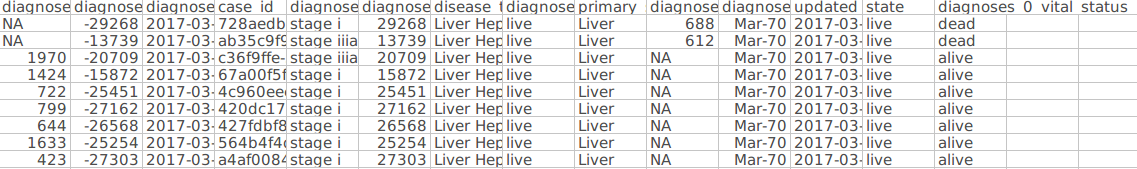
373个cases的注释
3. 根据file id使用TCGA 的API提取每个file的tsv格式的样本信息(主要是样本的类型:tumor还是normal),获得文件files_425_diagnoese.tsv:
#根据file ID创建过滤条件文件:
cat > ../payload <<EOF
{
"filters":{
"op":"in",
"content":{
"field":"files.file_id",
"value":[
EOF
# 将case ID写入条件:
row_num=`wc -l ../miRNA_caseID_fileID | awk '{print $1}' -`
awk -v row_num=$row_num '{if(NR<row_num)printf "\"%s\",\n",$3; if(NR==row_num)printf "\"%s\"\n",$3}' ../miRNA_caseID_fileID >> ../payload
# 创建条件结尾:
cat >> ../payload <<EOF
]
}
},
"format":"TSV",
"fields":"file_id,file_name,cases.submitter_id,cases.case_id,data_category,data_type,cases.samples.tumor_descriptor,cases.samples.tissue_type,cases.samples.sample_type,cases.samples.submitter_id,cases.samples.sample_id,cases.samples.portions.analytes.aliquots.aliquot_id,cases.samples.portions.analytes.aliquots.submitter_id",
"size":"10000",
"pretty":"true"
}
EOF
# 如果是用第三列的file ID,过滤条件里的field要修改为file_id_raw,
curl --request POST --header "Content-Type: application/json" --data @../payload 'https://api.gdc.cancer.gov/files' >../files_425_diagnoese.tsv
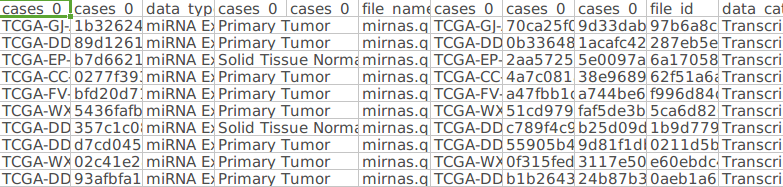
425样本的注释
从上面已经获得了肝癌组织全部的miRNA表达谱数据及临床数据,接下来就可以提取想要信息进行下一步分析,其他肿瘤的基因表达谱及临床数据下载与此类似,下次介绍TCGA数据整合与分析.
 苏公网安备 32050602011302号
苏公网安备 32050602011302号