摘要:
复现一篇nature cancer中的一个小图,很简单,不过如果不太熟悉ggplot和R的多图拼接的话,也着实需要花一番功夫。
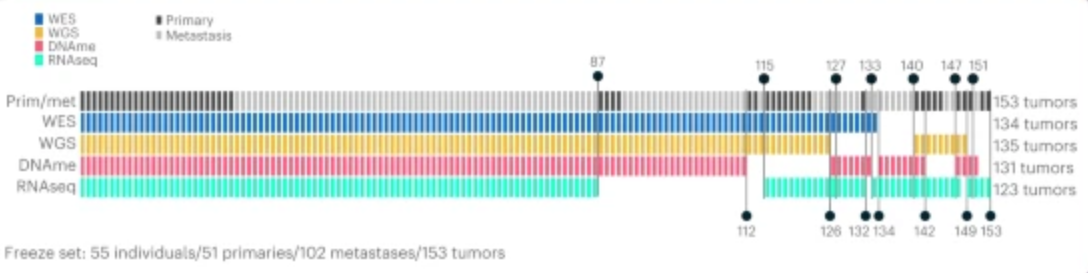
这个图就是给读者展示一下研究队列的一些基本信息,比方说我的队列中一共有多少病人,分别进行了什么组学分析。
我们假设我们的队列信息表是这样的,1代表有,0代表无。而在癌症类型中,1代表原位瘤,0代表转移瘤。大概如下所示:
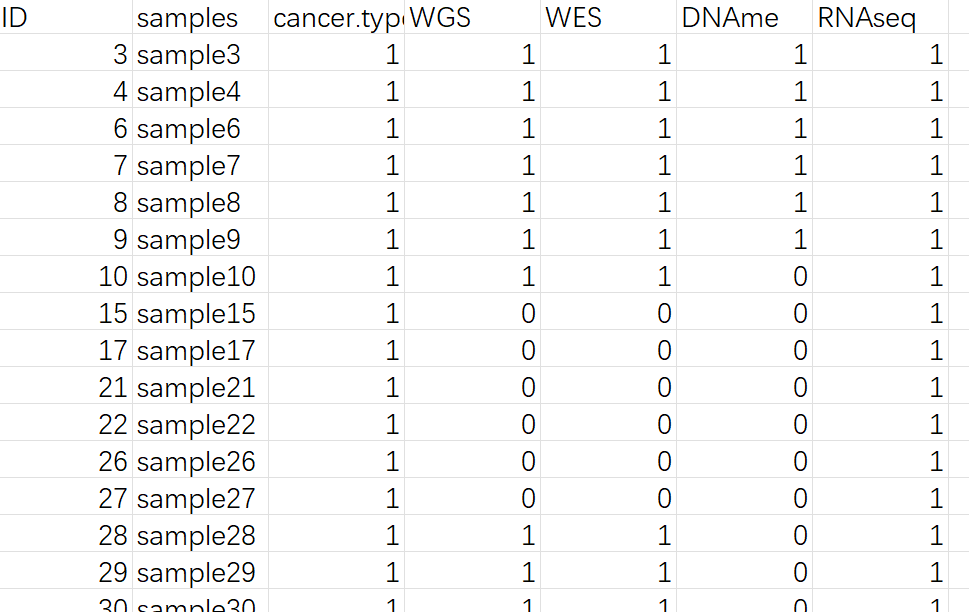
那么根据这个表,我们该如何做一个跟上面类似的图反映队列的信息呢?
如果你不想折腾的话,那么最简单的就是热图+ppt/ps。比方说使用pheatmap,你大概能得到这样的图:
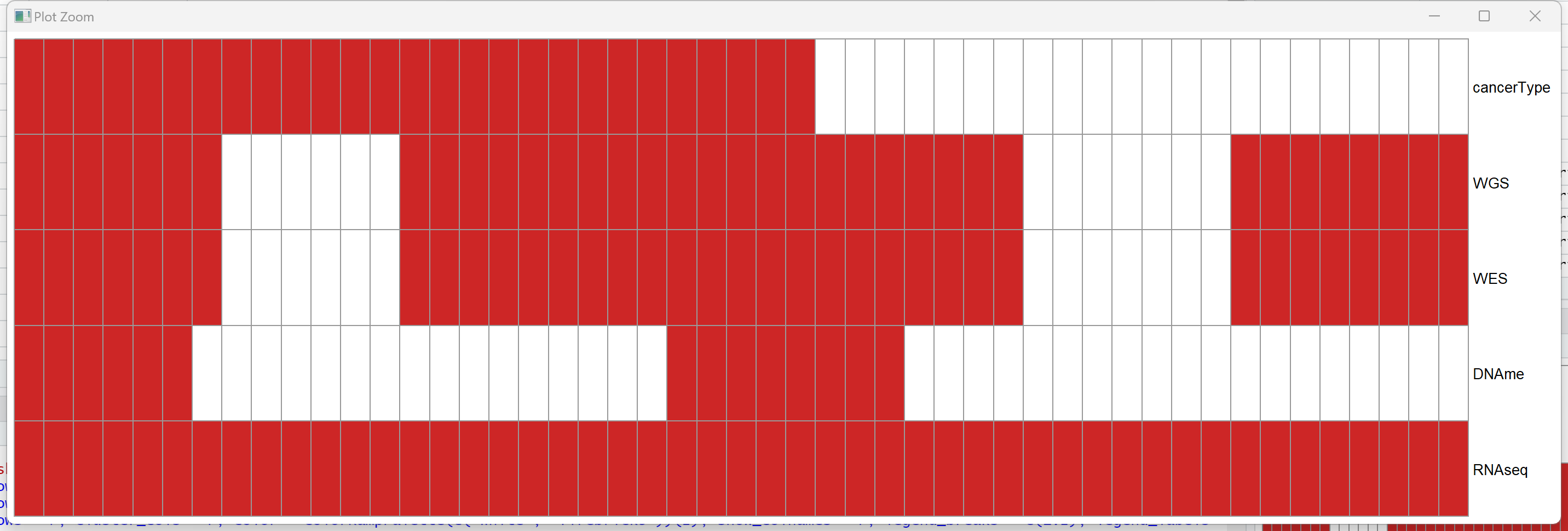
实现代码也就一行:
pheatmap(<你的数据框>, cluster_rows = F, cluster_cols = F, color = colorRampPalette(c("white", "firebrick3"))(2), show_colnames = F, legend = F)
不过使用热图来实现的话,其实有很多限制。一是不好控制颜色(像上面这样都是同样的颜色,其实挺丑的),字体等等。二是如果要添加删除信息啥的也很麻烦。那我们不如就来试试手动画图?
手动画图我们就得依赖ggplot了(功能实在太全了)。大概思路就是这样,把每一列数据单独做条形图,最后将条形图拼接起来即可。我这里就不做过多解释了,直接上代码:
#注意,代码得结合上面的表来看。如果你的信息表不是上面那样,需要将代码提取数据那部分改一下以适配你的数据表。
library(ggplot2)
library(lemon)
library(ggpubr)
library(gridExtra)
windowsFonts(CG=windowsFont('Century Gothic'))
rd<-read.csv('test.csv', header = T)
names(rd)<-c('ID','samples','cancerType','WGS','WES','DNAme','RNAseq')
ct<-rd[,c(1,2,3)]
wgs<-rd[,c(1,2,4)]
wes<-rd[,c(1,2,5)]
DNAme<-rd[,c(1,2,6)]
RNAseq<-rd[,c(1,2,7)]
p1<-ggplot(data = wgs, aes(x=ID, y = 1, fill = factor(WGS))) +
theme_classic() +
theme(axis.text.x = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.y = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank(),
axis.ticks.length = unit(0, "cm"),
plot.margin = unit(c(0,0,0,0), "cm"),
legend.position="none"
) +
geom_bar(stat = 'identity', width = 0.6) +
guides(fill=guide_legend(title=NULL)) +
scale_fill_manual(values = c('white','#F3B14F'), labels = c('', 'WGS')) +
geom_text(x = 0, y = 0.5, inherit.aes = FALSE, label = "WGS", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
geom_text(x = 65, y = 0.5, inherit.aes = FALSE, label = "xxx Samples", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
xlim(-10, 70)
p2<-ggplot(data = wes, aes(x=ID, y = 1, fill = factor(WES))) +
theme_classic() +
theme(axis.text.x = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.y = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank(),
axis.ticks.length = unit(0, "cm"),
plot.margin=unit(c(0,0,0,0), "cm"),
legend.position="none"
) +
geom_bar(stat = 'identity', width = 0.6) +
guides(fill=guide_legend(title=NULL)) +
scale_fill_manual(values = c('white','#176AB8'), labels = c('', 'WES')) +
geom_text(x = 0, y = 0.5, inherit.aes = FALSE, label = "WES", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
geom_text(x = 65, y = 0.5, inherit.aes = FALSE, label = "xxx Samples", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
xlim(-10, 70)
p3<-ggplot(data = DNAme, aes(x=ID, y = DNAme, fill = factor(DNAme))) +
theme_classic() +
theme(axis.text.x = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.y = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank(),
axis.ticks.length = unit(0, "cm"),
plot.margin=unit(c(0,0,0,0), "cm"),
legend.position="none"
) +
geom_bar(stat = 'identity', width = 0.6) +
guides(fill=guide_legend(title=NULL)) +
scale_fill_manual(values = c('white','#FC587A'), labels = c('', 'DNAme')) +
geom_text(x = 0, y = 0.5, inherit.aes = FALSE, label = "DNAme", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
geom_text(x = 65, y = 0.5, inherit.aes = FALSE, label = "xxx Samples", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
xlim(-10, 70)
p4<-ggplot(data = RNAseq, aes(x=ID, y = 1, fill = factor(RNAseq))) +
theme_classic() +
theme(axis.text.x = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.y = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank(),
axis.ticks.length = unit(0, "cm"),
plot.margin=unit(c(0,0,0,0), "cm"),
legend.position="none"
) +
geom_bar(stat = 'identity', width = 0.6) +
guides(fill=guide_legend(title=NULL)) +
scale_fill_manual(values = c('white','#30FEC5'),labels = c('', 'RNAseq'), limits = factor(c(0,1))) +
geom_text(x = 0, y = 0.5, inherit.aes = FALSE, label = "RNAseq", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
geom_text(x = 65, y = 0.5, inherit.aes = FALSE, label = "xxx Samples", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
xlim(-10, 70)
p5<-ggplot(data = ct, aes(x=ID, y = 1, fill = factor(cancerType))) +
theme_classic() +
theme(axis.text.x = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.y = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank(),
axis.ticks.length = unit(0, "cm"),
plot.margin=unit(c(0,0,0,0), "cm"),
legend.position="none"
) +
geom_bar(stat = 'identity', width = 0.6) +
guides(fill=guide_legend(title=NULL)) +
scale_fill_manual(values = c('#464646','#BBBBBB'), labels = c('metastasis', 'primary')) +
geom_text(x = 0, y = 0.5, inherit.aes = FALSE, label = "Prim/met", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
geom_text(x = 65, y = 0.5, inherit.aes = FALSE, label = "xxx Samples", check_overlap = TRUE, hjust = 1, size = 10, color = '#626262', family = 'CG') +
xlim(-10, 70)
'''
注意这里的逻辑:
1. 需要先将上述5个作图代码中的legend.position = "none"注释掉,然后p1-p5跑一遍。
2. 跑完后通过下面的get_legend()依次获取每个图的图例。并通过grid.arrange合并图例,然后再将legend.position = "none"恢复。
3. 恢复后再次执行p1-p5,然后依然通过grid.arrange来合并。
4. 最后将合并的图例和合并的条形图再整合,完成!
'''
lp1<-get_legend(p1)
lp2<-get_legend(p2)
lp3<-get_legend(p3)
lp4<-get_legend(p4)
lp5<-get_legend(p5)
lgs<-grid.arrange(lp5,lp2,lp1,lp3,lp4, ncol = 5, nrow = 1)
g1<-grid.arrange(p5,p2,p1,p3,p4, ncol = 1, nrow = 5)
png('test.png', units = 'in', width = 14, height = 3, res = 300)
grid.arrange(lgs, g1, ncol = 1, nrow = 2, heights = c(0.8, 2.8))
dev.off()
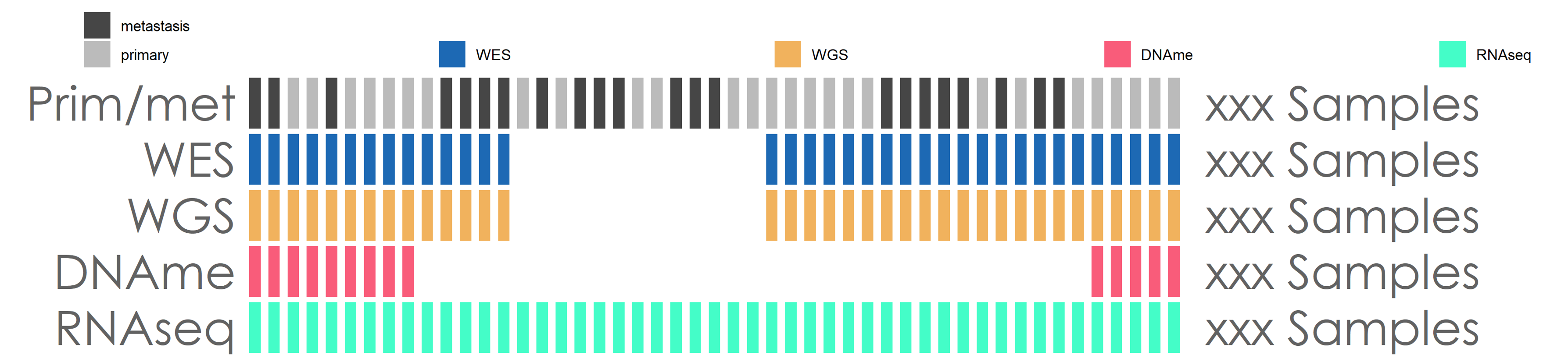
这段代码还是很丑的,更简单的方法就是直接写个函数,省的一段代码复制好几遍。Anyway,能完成任务就行,哈哈哈,反正只是画图,也不是搭应用,所以不需要考虑性能。
最终效果大概是这个样子:
2023.04.06更新:
今天在看文献时看到类似的图,作者使用的OncoPrint包实现了同样的效果。我没怎么用过这个包,印象中我只有画突变的瀑布图的时候用过它。如果有朋友对该包感兴趣可以自行百度相关教程,或者参考一下这个博文也行:《R 数据可视化 —— 聚类热图 ComplexHeatmap(五)oncoprint》
 苏公网安备 32050602011302号
苏公网安备 32050602011302号